Docking
The Docking package is a plugin that seamlessly integrates the AutoDock GPU utility with the Datagrok platform.
Getting started
Setting up docking configurations can be challenging, but we've streamlined this process. In this section, we'll cover everything from preparing configurations to storing them.
Prepare macromolecule (target)
Autodock plugin contains several pre-created targets. To add your own:
- Prepare the macromolecule and AutoDock grid parameter file using AutoDock tools.
- Ensure that the macromolecule is in the
PDBQTformat.
For detailed instructions, review the Autodock tutorials:
Put these files to the folder under System:AppData/Docking/targets. The name of the folder will appear as the target name in the Datagrok plugin UI.
Atomic maps:
The AutoDock calculation fails if for a particular ligand corresponding atomic maps are not available. To run docking on a big ligand dataset, we suggest including all available atomic map types:
A C HD N NA OA SA CL.
Prepare data
Create or load a dataframe that contains ligands to be docked. For demonstration purposes, consider using the provided demo data.
Run docking simulations
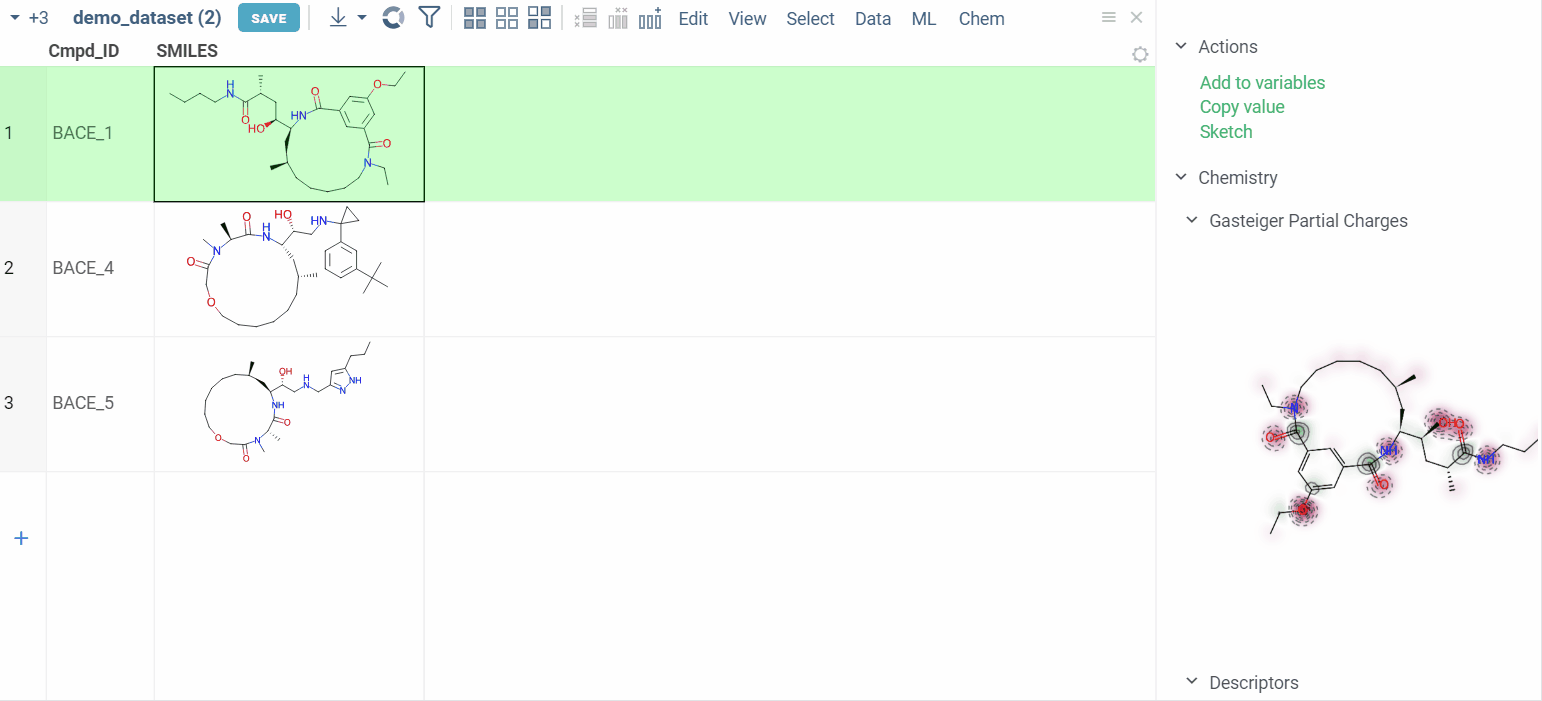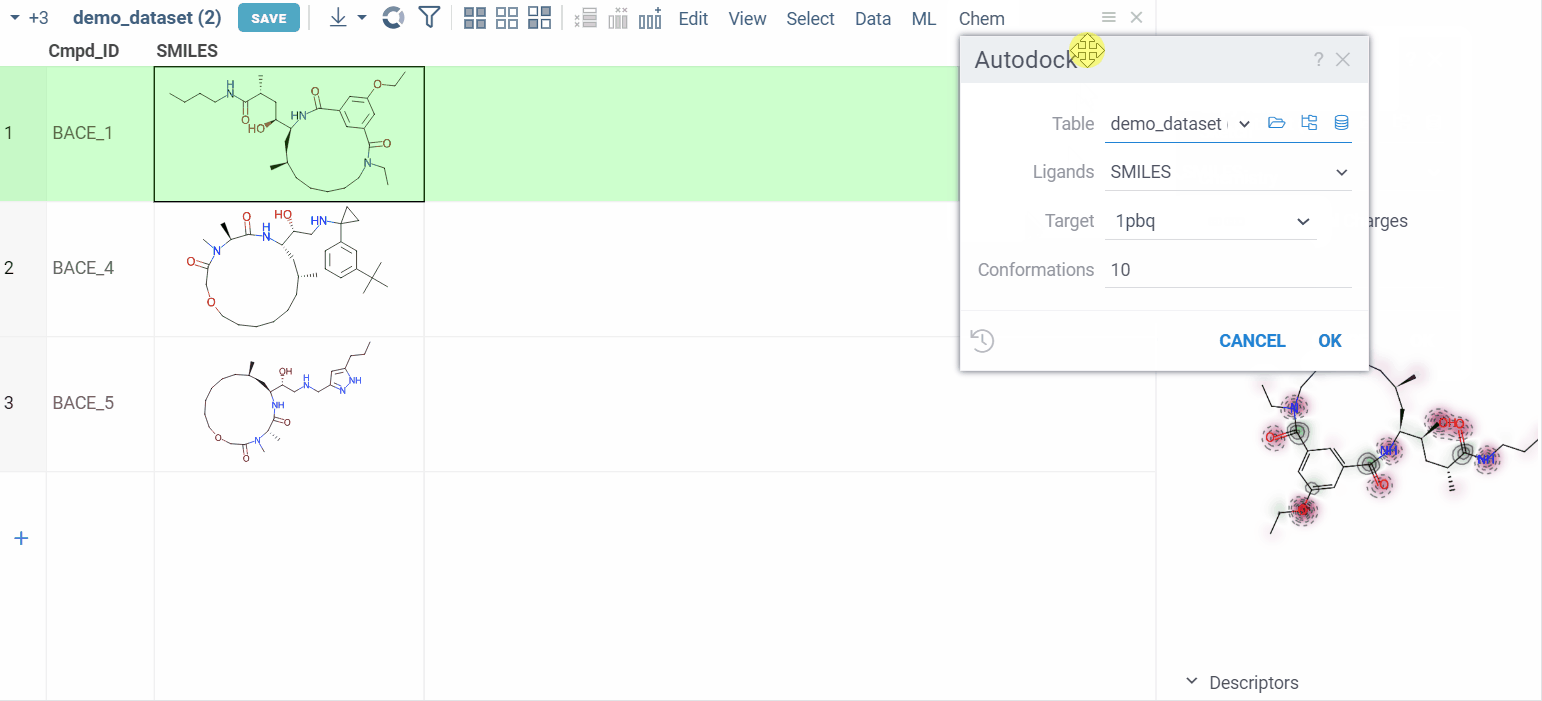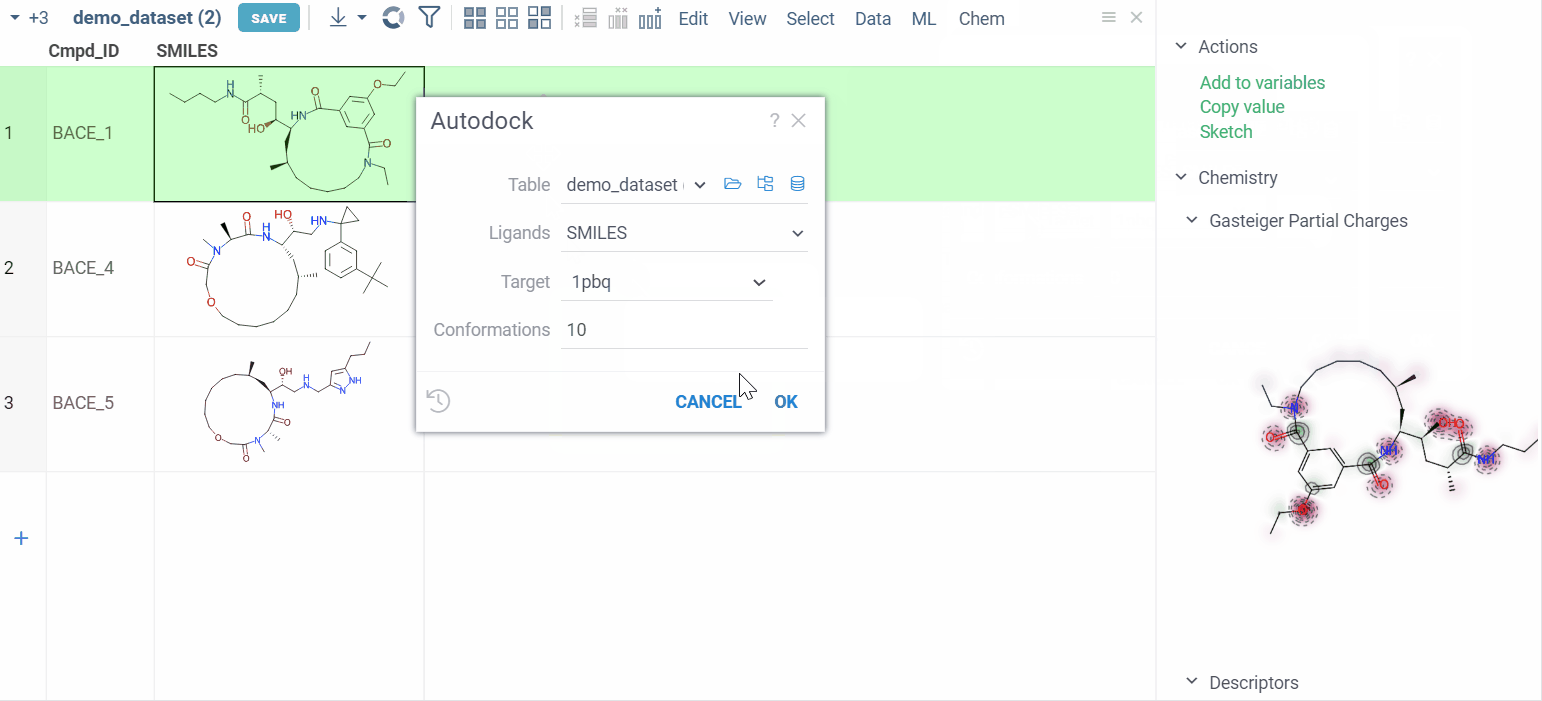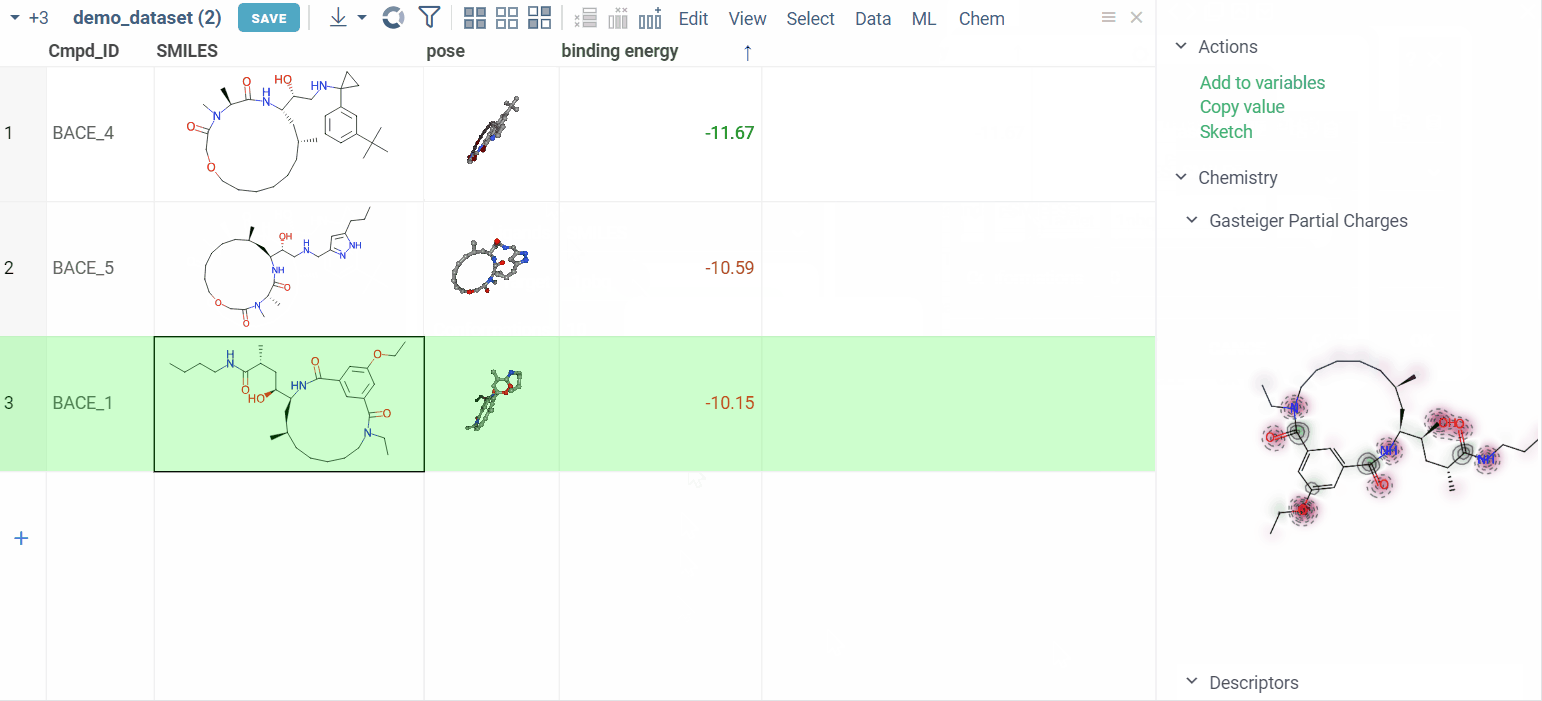
- Navigate to Chem > Autodock. A dialog appears.
- Configure the parameters:
- Ligands: Specify the column that contains small molecules to be docked.
- Target: Choose the folder that contains the macromolecule and grid parameter file.
- Conformations: Define the number of conformations.
- Run the calculations.
Performance note:
During the first run for a target Autodock calculates the macromolecule grids. Grid calculation works on a single CPU core, so it can take about a minute. Datagrok caches calculated grids, so subsequent runs with the same target will be much faster.
Features
After docking simulations are finished, various features become available. This section will guide you through them.
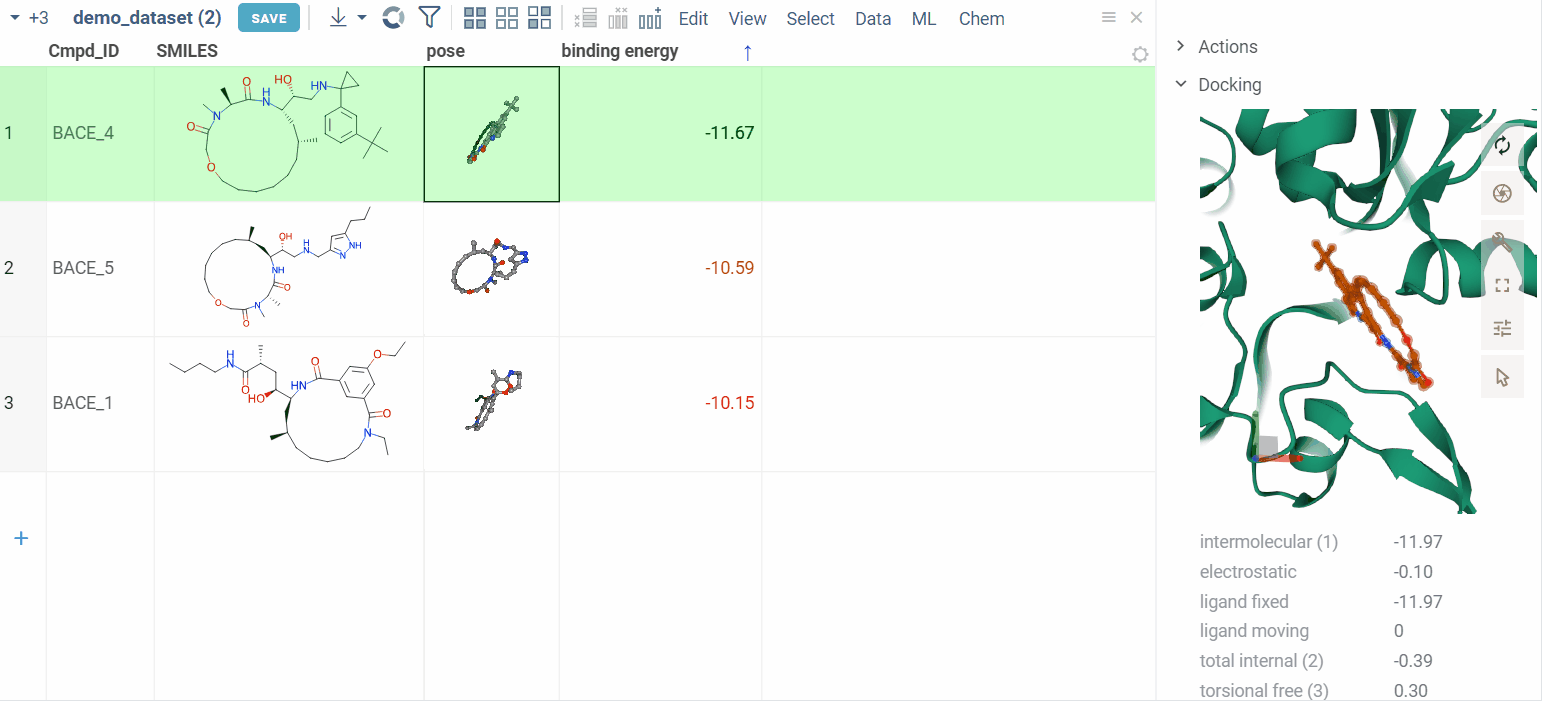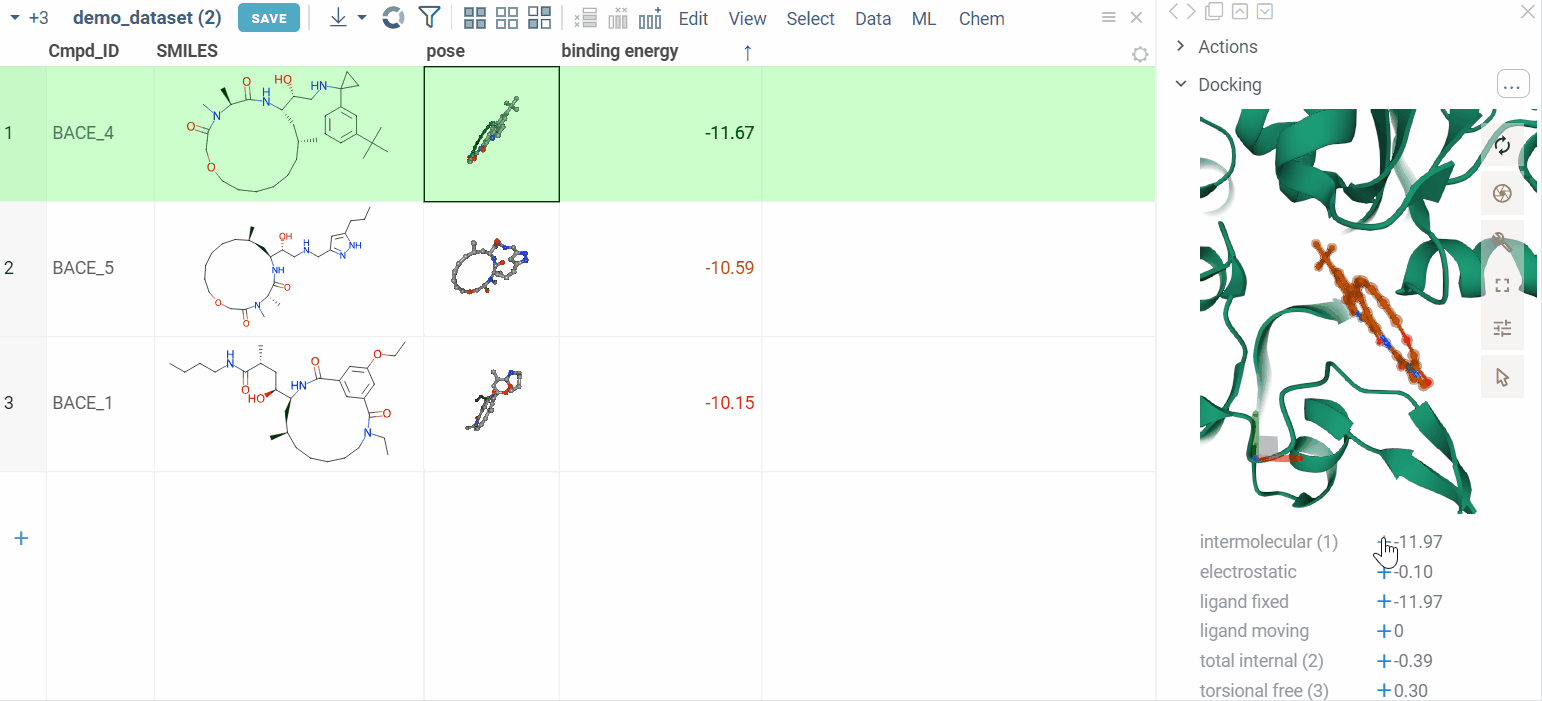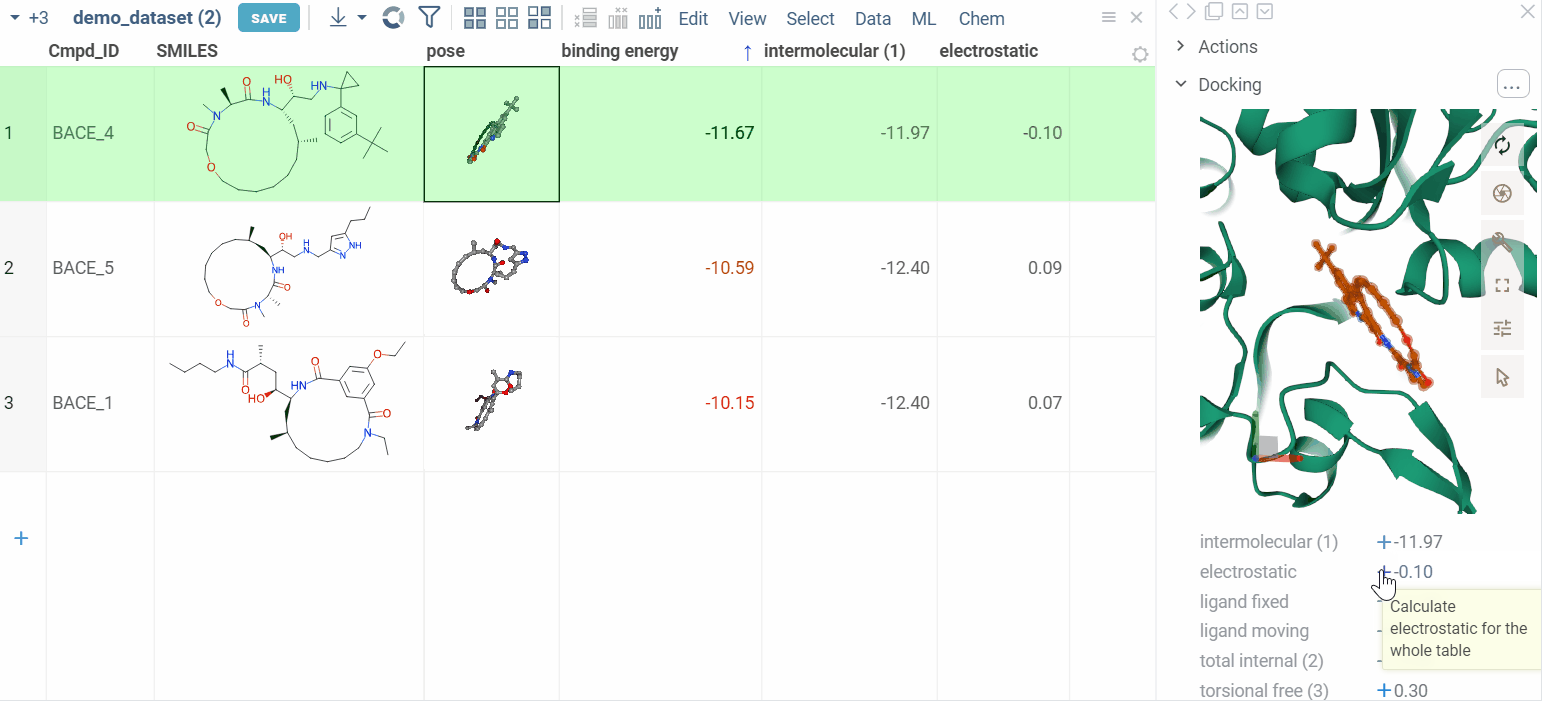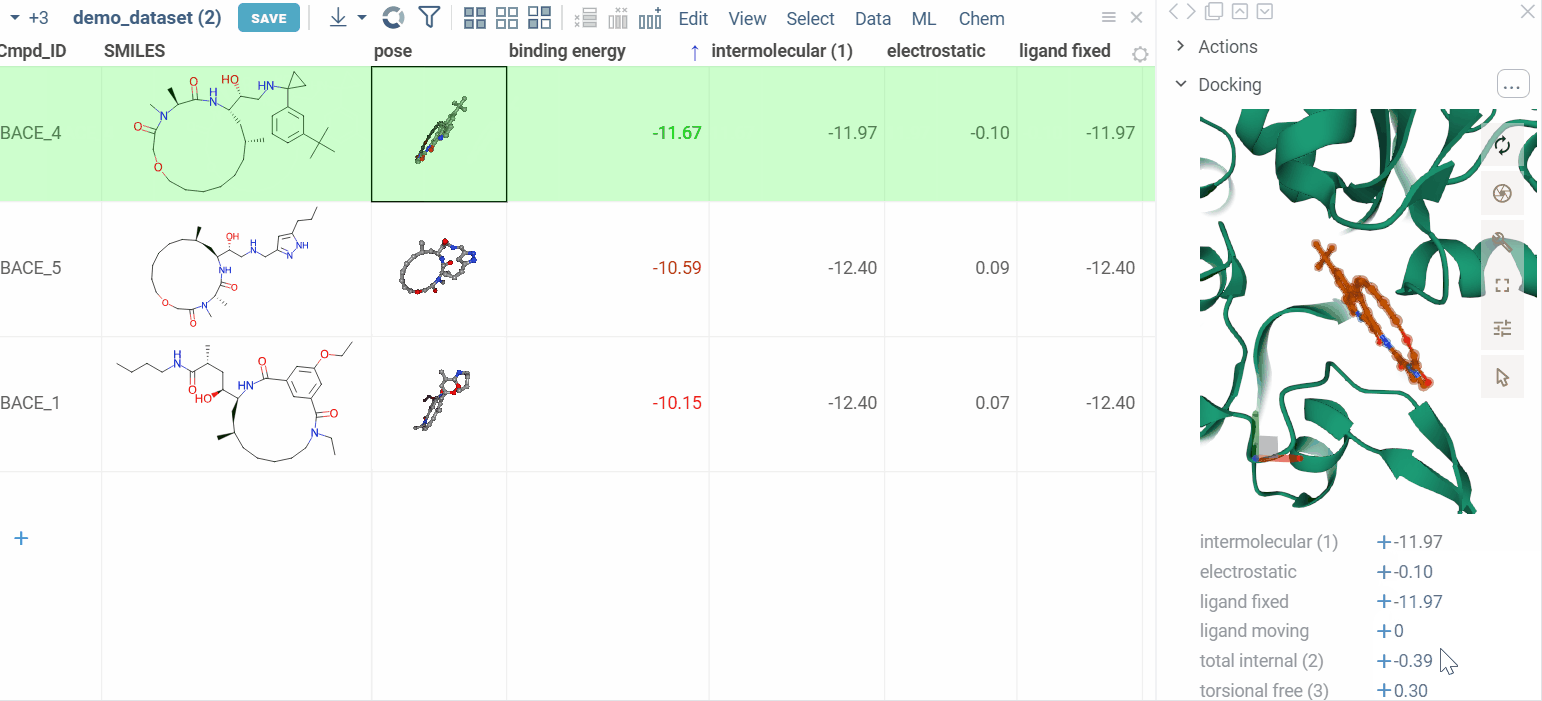
View docking results
Users can review the results: docking poses and binding energy values.
The column for binding energy shows how strong the connections are between ligands and receptors. Green indicates strong connections with low numbers, while red indicates weaker connections with higher numbers.
.default})
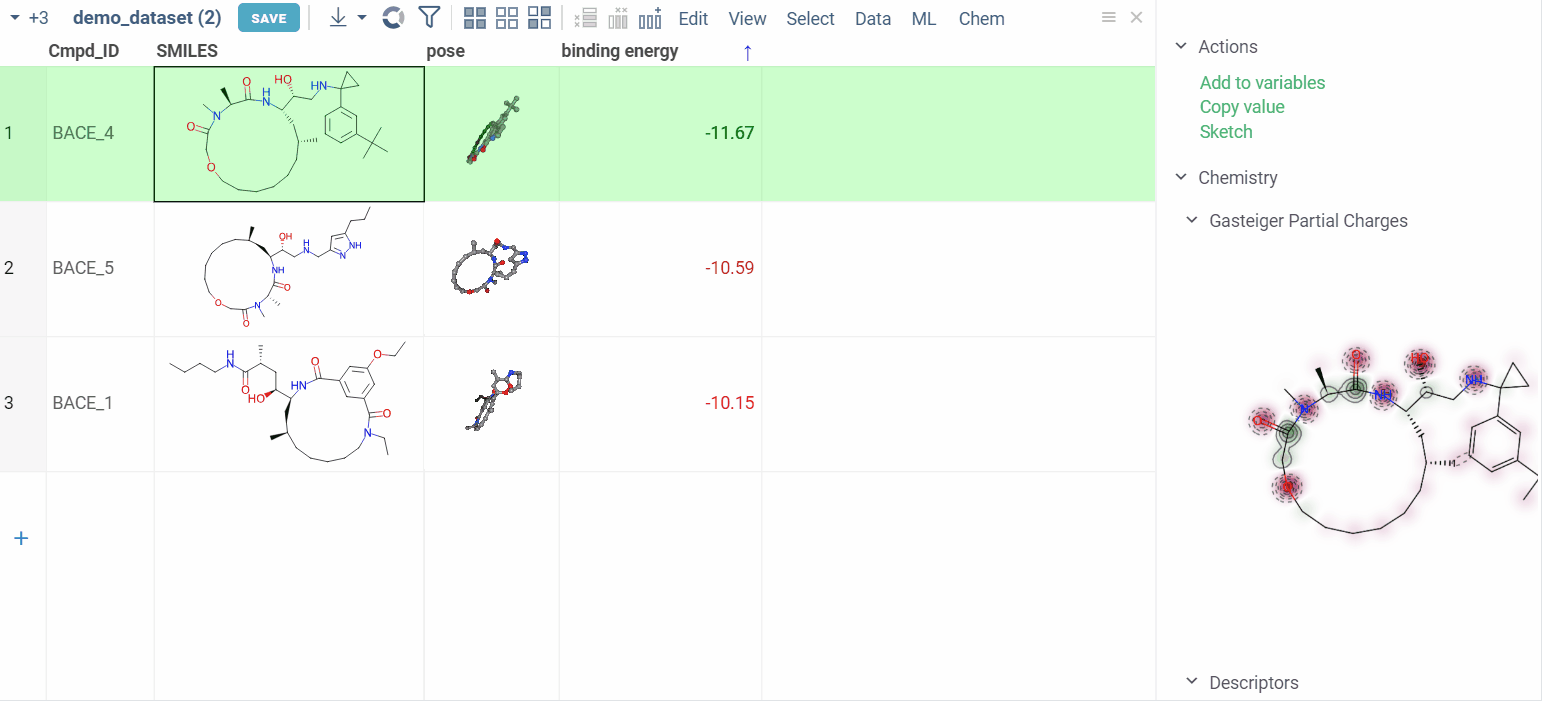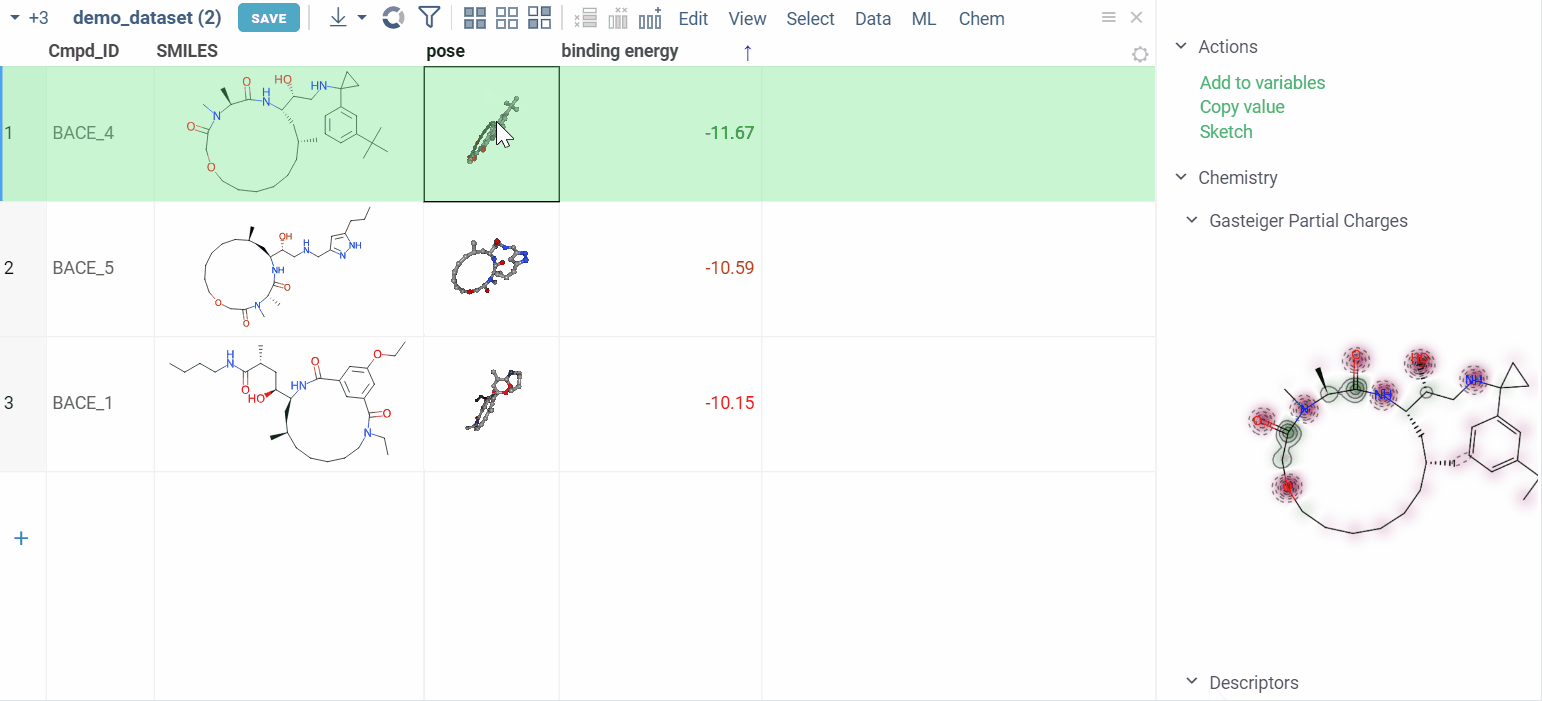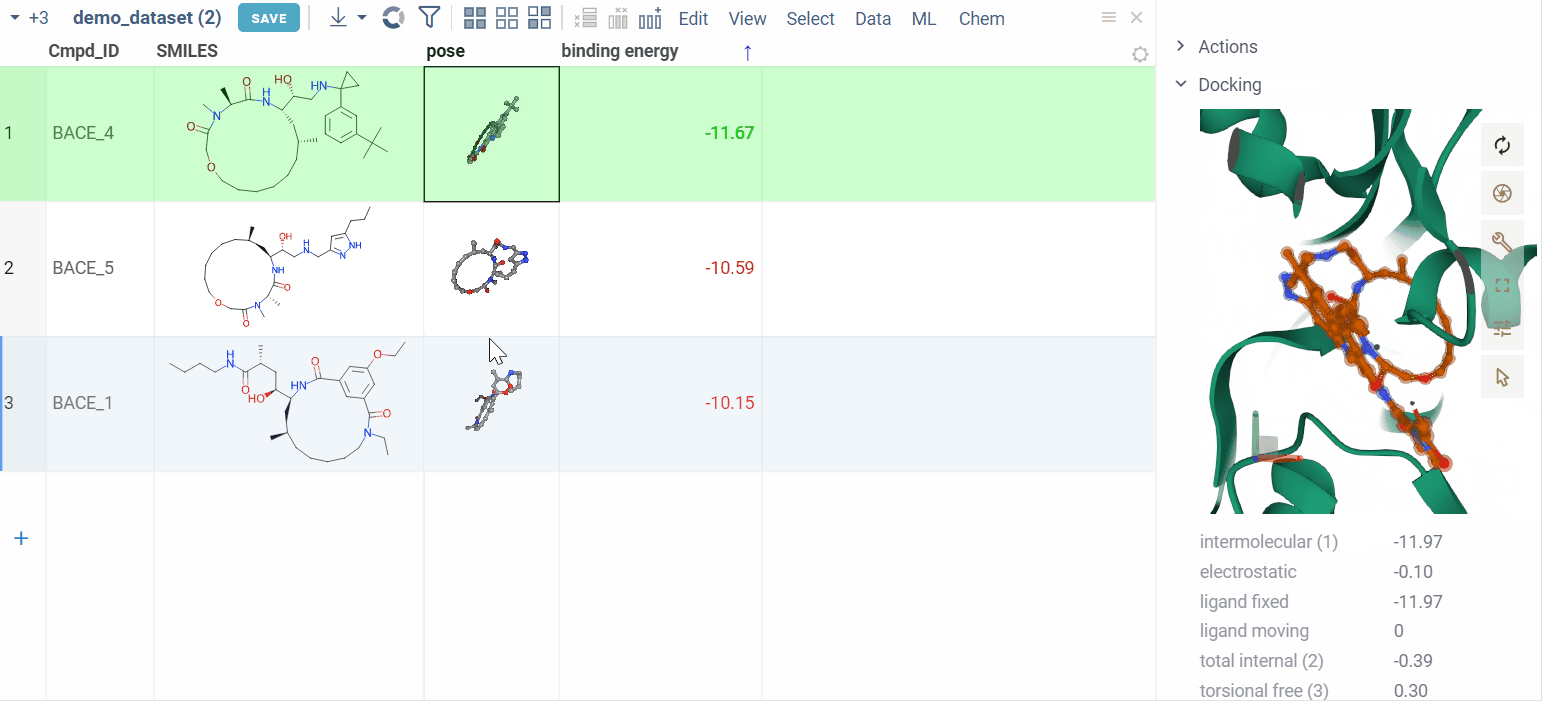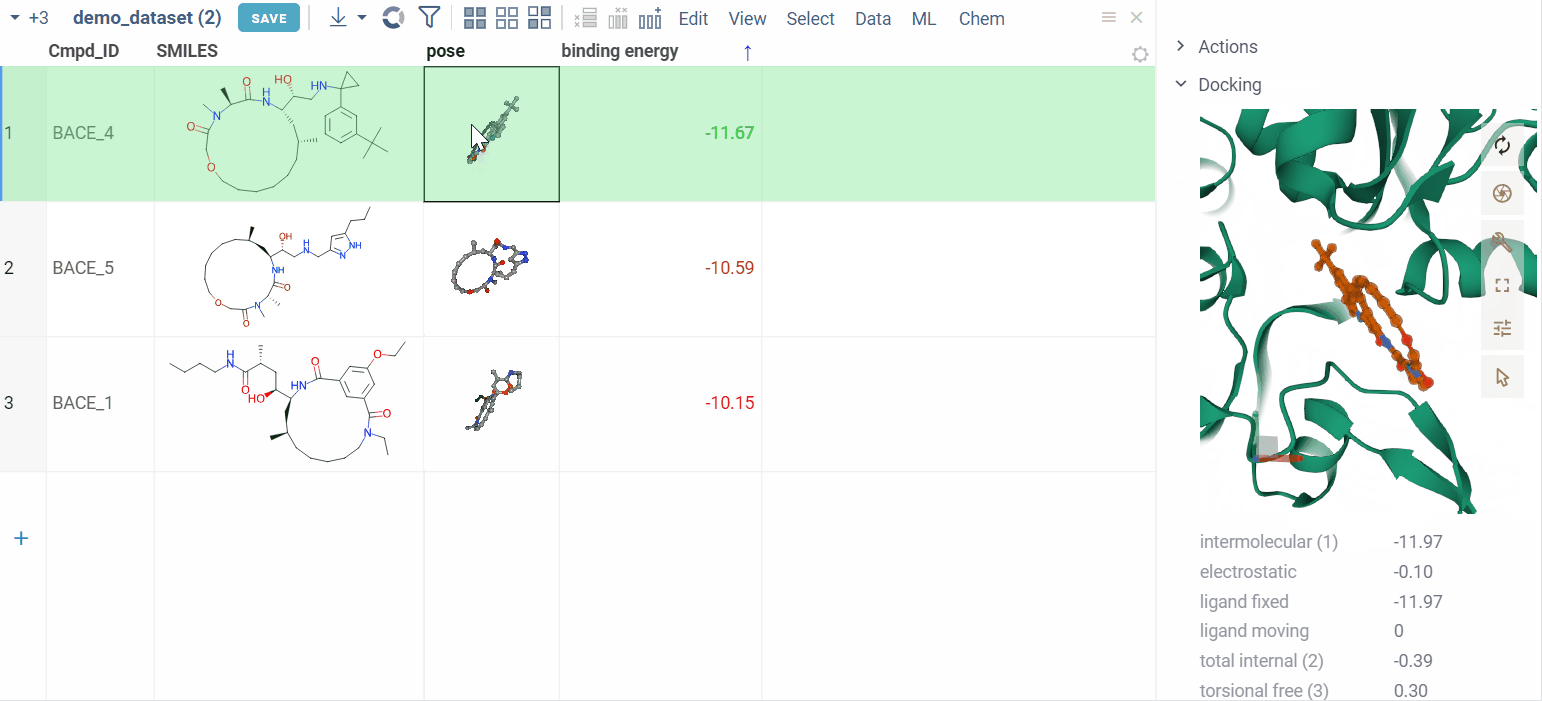
Zoom into binding pocket
Clicking on a pose activates a Molstar viewer in the context panel. It automatically zooms to the binding pocket.
Explore additional properties
We also show additional Autodock properties. Click the plus icon to add them to the whole dataframe.
Download pose in PDB and CIF formats
To download the pose within the protein context, right-click to open the menu. Choose "Download" and then select either as CIF or as PDB.